
Rational construction of polycystine-based nanoparticles for biomedical applications
 b
Jianxun
Ding
*ac
b
Jianxun
Ding
*ac
Abstract
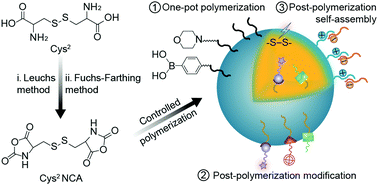
Polypeptide-based nanoparticles are one of the promising excipients of nanomedicines due to their excellent biosafety and flexible modification. Among all the types of polypeptide nanoparticles, polycystine (PCys2)-based ones draw increasing attention due to their unique properties. On the one hand, the uniformed nanogels can be easily obtained through the crosslinking of two active centers during polymerization without additional step of self-assembly. On the other hand, the Cys2-based nanoparticles always showed reduction-responsiveness owing to the inherent disulfide bond. With the development of advanced diagnostic and therapeutic technologies, the multi-functional PCys2-based nanoparticles were achieved via rational construction of the polymer structure. This review summarizes the overall development of Cys2-based polypeptide nanoparticles, especially the structural design for the generation of multi-functional nanoparticles, along with their corresponding biomedical applications.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

