
Zwitterionic choline phosphate conjugated folate-poly (ethylene glycol): a general decoration of erythrocyte membrane-coated nanoparticles for enhanced tumor-targeting drug delivery†

Abstract
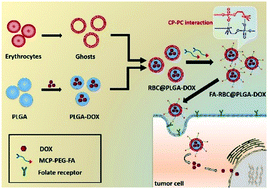
Erythrocyte membrane nanosystems have become one of the important research directions of disease treatment, especially for tumor treatment, and can enhance the long circulation time of anti-cancer drugs in vivo, and penetrate and accumulate in the tumor site effectively. However, erythrocyte membranes lack targeting properties and it is necessary to provide tumor-targeting function by modifying erythrocyte membranes. In this study, we report on a novel modification method of an erythrocyte membrane nanosystem to target tumors. Specifically, the tumor-targeting molecule folate-poly (ethylene glycol) (FA-PEG) was modified with a zwitterionic 2-(methyl acryloyoxy) ethyl choline phosphate (MCP) by the Michael addition reaction to obtain MCP-modified FA-PEG (MCP-PEG-FA). Based on the strong “N–P” tetravalent electrostatic interaction between MCP and phosphatidyl choline on the erythrocyte membranes, MCP-PEG-FA can be modified on the erythrocyte membrane encapsulated doxorubicin (DOX) loaded poly(lactic-co-glycolic acid) (PLGA) nanosystem to form a tumor-targeting erythrocyte membrane nanosystem (FA-RBC@PLGA-DOX). The results show that MCP-PEG-FA was synthesized and successfully bonded to the erythrocyte membrane nanosystem, and the FA-RBC@PLGA-DOX nanosystem had a better tumor-targeting function and tumor killing effect compared with those of the nanosystems without FA ligand modification. The universal modification method of erythrocyte membranes is successfully provided and can be applied to the treatment of various diseases.
- This article is part of the themed collection: Bioinspired Surfaces Engineering for Biomaterials
 Please wait while we load your content...
Please wait while we load your content...

