
Recent advancement in heterogeneous CO2 reduction processes in aqueous electrolyte

Abstract
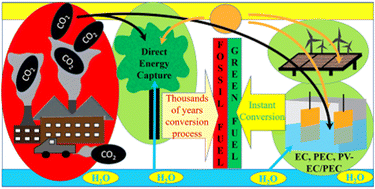
Energy is essential in the human race toward prosperity and it always remains in peak demand. Conventional fossil fuels may fulfil this energy demand for now but at the cost of severe long-term adverse effects on the environment. Therefore, many research groups are working to develop efficient and affordable processes for green energy harvesting to ensure true prosperity and a healthy environment for future generations. In this context, the CO2 reduction reaction (CO2RR) is emerging as one of the most essential and potent research fields for sustainable zero-emission green energy harvesting. Although there are a lot of reviews on the CO2RR, there is a lack of systematic comparisons of various common electro or non-electro catalytic processes, discussion of progress over time, and in-depth analysis with respect to materials. This review briefly introduces numerous heterogeneous catalytic CO2RR processes, categorized based on fundamental differences, and provides an up-to-date overview of catalyst materials and improvements in catalysis setup. In this review, electrochemical (EC), photoelectrochemical (PEC), and photovoltaic cell-assisted EC/PEC (PV-EC/PEC) processes with several modifications have been emphasized, including a detailed overview of the futuristic membrane electrode assembly (MEA) method, and they are comprehensively discussed following a timeline format. Along with the underlying comparison of various processes, strategies for the further utilization of these processes/materials and challenges for future work have also been summarized.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

