
Recent strategies for activating the basal planes of transition metal dichalcogenides towards hydrogen production
 *a
*a
Abstract
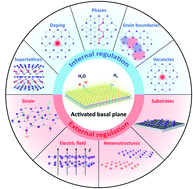
The hydrogen evolution reaction (HER) presents an environmentally friendly, efficient, and cost-effective way to produce hydrogen fuel. Recently, two-dimensional (2D) transition metal dichalcogenides (TMDCs) have emerged as a fascinating class of HER materials. Their atomically thin nature acts as an interface with the possibility of maximum exposure of nearly all active sites to the reaction environment. However, for most non-metallic TMDCs, as exemplified by 2H-MoS2, 1T-PtSe2, and 1T′-ReSe2, their basal planes are usually HER-inert except for minor edges. Based on their rich chemistry, significant progress has been recently made to activate the basal planes for HER. This review provides an overview of the current activating strategies. They can be classified into internal and external regulations, depending on whether the pristine structure is altered or not. The former directly creates more active sites through doping, vacancies, phases, grain boundaries (GBs), and superlattices. On the other hand, the latter indirectly promotes the activity utilizing external methods such as the electric field, strain, substrates, and heterostructures. We discuss each strategy's principles and characteristics and highlight their enhanced catalytic activities. We finally provide personal perspectives on the challenges and opportunities in this emerging field, including a joint strategy, a phase-engineering strategy, electrical–electrocatalytic coupling, as well as the potential of “single-atom-layer” catalysis.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry A Lunar New Year collection, 2022 Journal of Materials Chemistry A Most Popular Articles and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

