
Approaching a stable oxygen redox reaction in lithium-rich cathode materials: structural perspectives from mechanism to optimization
 *acde
*acde
Abstract
Oxygen redox (OR) chemistry has been an attractive topic in the field of high-energy lithium-ion batteries, as it enables extra storage of charge and boosts the capacity of highly potential layered Li-rich oxide (LLO) cathode materials. However, the OR reaction is usually irreversible during the electrochemical process, inducing severe performance degradation that sets an impenetrable barrier to the LLO applications. Over the last two decades, great efforts have been made to fundamentally understand the irreversibility of OR, finally reaching a consensus that it is deeply rooted in the structural features of LLOs. Although the structural mechanism is complex and still remains to be further clarified, the current findings of the structure–OR coupling have already inspired blooming optimistic expectations from structural perspectives. Herein, we systematically review the recent progress of the OR investigations in LLOs, with a special emphasis on deciphering the structure–OR coupling. Moreover, efficient structural control strategies for promoting the reversibility of OR are also introduced, followed by an outlook on future rational design and development of LLO materials. This comprehensive summary and perspective are expected to be helpful to promote further OR and LLO research.
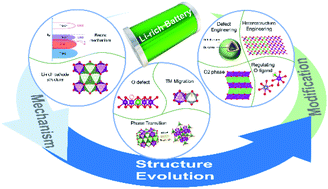
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

