
Quasi-2D halide perovskite crystals and their optoelectronic applications
 *b
*b
Abstract
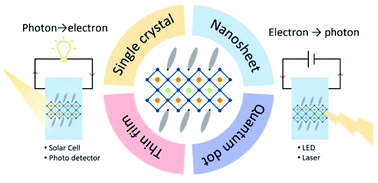
Due to the widely tunable and exceptional semiconductor properties, quasi-two-dimensional (2D) halide perovskites have been considered outstanding candidates for next-generation optoelectronic devices and have thus undergone rapid development over the last decade. Their property tunability is intimately correlated with the quantum-well thickness (corresponding to the n value, the number of octahedral layers in each layer of quasi-2D halide perovskites). However, a systematic review of the relationship between the n value and the optoelectronic properties/applications of the quasi-2D perovskites is lacking. In this review, we begin with the n-dependent properties of quasi-2D perovskites, and move on to the synthesis of different forms of quasi-2D perovskite crystals. Afterwards, we focus on the existing optoelectronic applications using quasi-2D perovskites including light-emitting diodes, solar cells, lasers, and photodetectors. In the last section, we discuss the current challenges and a few promising directions with a special focus on the precise control of the n value. Moreover, the possible non-centrosymmetrical ordering of the organic ligands in quasi-2D halide perovskites provides new insights and opportunities to study the interaction between spontaneous polarization, photovoltaic effect, and carrier transport behaviors in the corresponding electronic and optoelectronic devices.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

