
Advances in understanding and regulation of sulfur conversion processes in metal–sulfur batteries
 d
and
Zheng-Long
Xu
*ace
d
and
Zheng-Long
Xu
*ace
Abstract
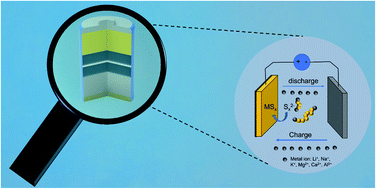
Lithium–sulfur batteries (LSBs) have attracted intensive attention as promising next-generation energy storage systems, due to the high energy density and low cost of sulfur cathodes. Despite the substantial progress in improving LSBs’ performance, their wide implementation still suffers from great challenges, including the difficulties in achieving practically high energy density with long cycle life and the concerns about the limited lithium resources. The former issue mainly arises from the insufficient understanding of the mechanics of the complex lithium–sulfur redox reactions, while the latter trigger the exploration of a range of new metal–sulfur systems, such as sodium–sulfur, potassium–sulfur, magnesium–sulfur, calcium–sulfur, and aluminum–sulfur batteries. These lithium-free metal–sulfur batteries (MSBs) have the potential to offer higher energy density or/and lower battery costs. The fundamental understanding and rational regulation of effective metal–sulfur conversion reactions are crucial for developing advanced and emerging MSBs. Herein, this work aims to overview the state-of-the-art progress in circumventing these issues of MSBs, in terms of working mechanisms, key factors determining the electrochemical behavior and battery performance. Advanced in situ characterization techniques used to disclose the sulfur conversion mechanisms are also elaborately discussed. Conclusions and perspectives for the future research direction in MSBs are proposed.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

