
Carbon nanomaterials–polymer composites for perovskite solar cells: preparation, properties and applications

Abstract
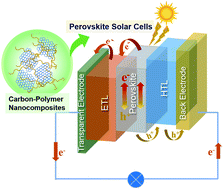
Perovskite solar cells (PSCs) are a rising star in the photovoltaic industry, which achieved an enormous breakthrough in terms of efficiency from an initial 3.8% in 2009 to 25.7% in 2021. The major challenge to bring perovskite solar cells commercially available is the poor long-term device stability. The defects in perovskite and energy loss originating from charge recombination limit the efficiency of perovskite solar cells. Carbon–polymer composites, which feature superb electrical conductivity, outstanding carrier mobility, remarkable flexibility and superior heat and moisture stability, have attracted considerable attention to tackle these issues. With their multifunctional properties, carbon–polymer composites can play various roles in almost every component in the perovskite solar cell architecture. In this review article, recent progress concerning the utilization of carbon–polymer composites in different components in PSCs (i.e., perovskite additives, electrodes, encapsulation layers and charge transport layers) is comprehensively overviewed. Then, future research directions and opportunities toward high-performance perovskite solar cells utilizing carbon–polymer composites are presented.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

