
Solvent effect on the Seebeck coefficient of Fe2+/Fe3+ hydrogel thermogalvanic cells†

Abstract
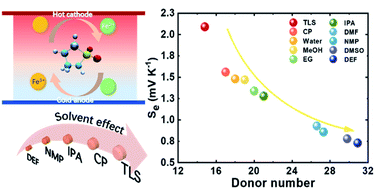
Thermogalvanic (TG) cells offer a clean and scalable energy conversion pathway for the recovery of low-grade waste heat. Enhancing the voltage generated by a single TG cell is crucial for widespread application in the internet of things (IoT) and wearable electronics. But the mechanism for manipulating the thermo-voltage is still unclear. In this work, we investigate the solvent effect on the TG performance by introducing nine organic solvents with different Gutmann donor numbers (DNs) into an aqueous Fe2+/Fe3+ electrolyte. A strong reverse correlation between the solvent DN and the Seebeck coefficient of the TG cell is observed. Among these, the tetramethylene sulfone–Fe2+/Fe3+ hydrogel TG cell exhibits a Seebeck value of 2.49 mV K−1, which is the largest reported value for Fe2+/Fe3+ based TG cells to date. A combination of experiments and molecular dynamics is used to elucidate the role of solvent DN on the Seebeck values. It is found that the difference in entropy contributions from changing solvation shell sizes as a result of organic solvent addition is the origin of the observed dependency between the Seebeck coefficient and solvent DN. This work provides a new perspective for the enhancement of TG performance, and this approach can be extended to other electrolyte systems and realize the application of TG cells.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry A Lunar New Year collection, 2022 Journal of Materials Chemistry A Most Popular Articles and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

