
Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: from nanoparticle to single atom catalyst
 *b
and
Chang-jun
Liu
*a
*b
and
Chang-jun
Liu
*a
Abstract
Supported Ni catalysts are promising for CO2 hydrogenation because of their relatively cheap price with comparable activity to noble metal catalysts. The product of CO2 hydrogenation over a supported Ni catalyst can theoretically be methane, CO, methanol and formic acid. The electronic and geometric structures of a supported Ni catalyst have a significant effect on the activity and selectivity of CO2 hydrogenation. Supported single nickel atom catalysts are found to tend to form carbon monoxide, methanol and, theoretically, formic acid. The selectivity depends on the support. The supported nickel cluster on indium oxide and In2O3–ZrO2 is highly selective for methanol synthesis. Supported nickel nanoparticles are normally good catalysts for methane formation at reasonably low temperatures. However, the structural effect of the supported Ni catalyst on CO2 hydrogenation has not been well investigated. The mechanism is still in debate. To further improve the activity and stability with tunable selectivity, the structure control of the Ni catalyst is very necessary for CO2 hydrogenation, which is highly structure sensitive. In this review, recent advances in the understanding of the structural effects of supported Ni catalysts on CO2 hydrogenation are summarized, including theoretical studies, operando or in situ catalyst characterization and experimental studies. Future development is therefore finally addressed.
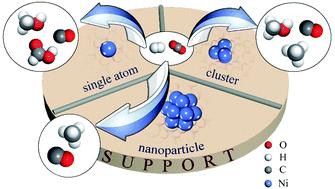
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

