
Surface engineering of anode materials for improving sodium-ion storage performance
 a
Xiu Song
Zhao
*a
a
Xiu Song
Zhao
*a
Abstract
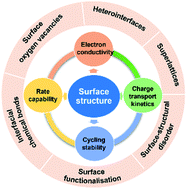
Sodium-ion batteries (NIBs) are considered a complementary or even an alternative energy storage technology to lithium-ion batteries (LIBs). Current technological development of NIBs is hindered by fundamental challenges, such as sluggish charge transport kinetics, short cycling lifespan, and low energy density. Electrode materials hold the key to any success in commercialising NIBs. While the past decade has witnessed rapid advances in design and synthesis of various electrode materials with different structures and microcosmic morphologies, there are still critical issues associated with electrode materials, especially anode materials, that need to be addressed before the NIB technology becomes commercially viable. For battery electrode materials, their surface properties play a critical role in determining cell performance. As a forefront of an electrode material where Na ion storage and charge transfer initiate, the electrode surface has a fundamental influence on the charge storage properties of the electrode. In this article, we review recent research progress towards modification/functionalisation of the surface structure of NIB anode materials for improving charge transport kinetics, charge storage capacity, and cycling durability. We aim to provide a concise summary of strategies for surface engineering, along with an insightful analysis of the critical role of the surface structure of NIB anode materials in determining their electrochemical performance.
- This article is part of the themed collection: Editor’s Choice: Beyond Li: Alternative battery chemistries
 Please wait while we load your content...
Please wait while we load your content...

