
Multiscale porous single-atom Co catalysts for epoxidation with O2†
 *a
and
*a
and
Abstract
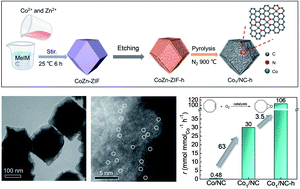
Epoxides are versatile intermediates in the production of a diverse set of chemical products. The direct epoxidation of alkenes using O2 represents an environmentally friendly and economical process to replace the use of expensive H2O2 or organic peroxides for the synthesis of epoxides. Herein, single-atom Co anchored on N-doped carbon supports with a multiscale porous structure (Co1/NC-h) has been successfully constructed by etching CoZn-ZIF metal–organic framework precursors before a carbonation process. Benefitting from the high intrinsic activity of Co–Nx active sites and the more accessibly exposed reactive sites of multiscale porous structures, the Co1/NC-h catalysts can achieve the epoxidation of cyclooctene with 95% yield of 1,2-epoxycycloheptane at 140 °C and 0.5 MPa O2. This research opens up opportunities for designing high-performance single-atom catalysts towards applications in diverse heterogeneous catalysis.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

