
Synergistically enhanced single-atomic site catalysts for clean energy conversion

Abstract
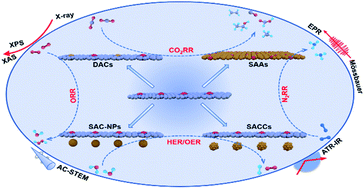
The development of cost-effective, high-performance catalysts at the atomic level has become a challenging issue for large-scale applications of renewable clean energy conversion. With adjustable structural characteristics and maximum atomic utilization efficiency, single-atomic site catalysts (SACs) are considered to be the most potential next-generation materials. Moreover, the introduction and coupling of some synergistic components is desirable to accurately regulate the structural interactions and lead to improved activity. In addition to the strong metal–support interaction (SMSI) in SACs, there is tremendous opportunity to explore and develop the synergistic effect of SAC-nanoparticles (SAC-NPs), SAC-clusters (SACCs), dual-atom site catalysts (DACs), and single-atomic alloys (SAAs). Moreover, these unique synergistic structures between adjacent atomic sites could still maintain their high atomically dispersed nature and stability. In this review, we begin by introducing the types, synthetic strategies and characterization methods of SAC-NPs, SACCs, DACs and SAAs, discussing the key factors controlling their structures. We then review several important clean energy catalytic reactions performed over these atomic-coupling structures, and compare the respective advantages and disadvantages of SACs, SAC-NPs, SACCs, DACs and SAAs. Finally, the challenges and perspectives of this unique single-atomic site synergistic effect are suggested. We believe that this critical review provides guidance for the rational design of new single-atomic site catalysts for clean energy conversion.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

