
Zinc/graphitic carbon nitride co-mediated dual-template synthesis of densely populated Fe–Nx-embedded 2D carbon nanosheets towards oxygen reduction reactions for Zn–air batteries†

Abstract
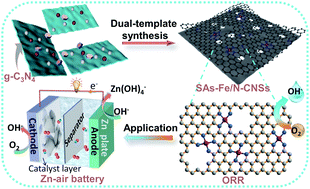
Atomically dispersed Fe–N–C catalysts have been extensively deemed as appealing substitutes for Pt-series catalysts towards oxygen reduction reactions (ORRs). Nevertheless, most reported Fe–N–C materials suffer from inefficient Fe-based nanoparticles and low-density Fe–Nx sites. Herein, a Zn/g-C3N4-mediated dual-template strategy was employed to synthesize densely populated atomic Fe–Nx center-embedded N-doped carbon nanosheets (SAs-Fe/N-CNSs) with adjustable porous structures by the simple pyrolysis of D-glucosamine/FeZn/g-C3N4 complexes. g-C3N4 works as a structure-guiding 2D template and offers abundant coordination-N trapping sites for anchoring Fe atoms, simultaneously. ZnCl2 serves as a self-sacrificial template creating a hierarchical porous structure by its volatilization as well as hinders the agglomeration of Fe atoms by spatial segregation during pyrolysis. Due to the high-density atomic Fe–Nx moieties, unique 2D structure, hierarchical porosity, and large surface area, the optimal SAs-Fe/N-CNS catalyst exhibits satisfying ORR performance including excellent activity (E1/2 = 0.91 V) and desirable durability, surpassing the Pt/C catalyst. Additionally, the superb performance of SAs-Fe/N-CNS-based Zn–air batteries with a maximum power density of 157.03 mW cm−2 verifies their promising application in practical electrochemical systems.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

