
A MnOx enhanced atomically dispersed iron–nitrogen–carbon catalyst for the oxygen reduction reaction†
 ‡a
Mingjie
Xu,
c
John
McCloy,
a
Guodong
Ding,
e
Qiang
Zhang,
e
Qiurong
Shi,
a
Dan
Du,
a
Jin-Cheng
Li,
*a
Xiao
Zhang
*f
and
Yuehe
Lin
*aef
‡a
Mingjie
Xu,
c
John
McCloy,
a
Guodong
Ding,
e
Qiang
Zhang,
e
Qiurong
Shi,
a
Dan
Du,
a
Jin-Cheng
Li,
*a
Xiao
Zhang
*f
and
Yuehe
Lin
*aef
Abstract
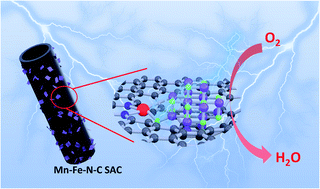
Cost-effective and highly efficient Fe–N–C single-atom catalysts (SACs) have been considered to be one of the most promising potential Pt substitutes for the cathodic oxygen reduction reaction (ORR) in proton exchange membrane fuel cells (PEMFCs). Nevertheless, they are subject to severe oxidative corrosion originating from the Fenton reaction, leading to poor long-time durability of PEMFCs. Herein, we propose a MnOx engineered Fe–N–C SAC (Mn–Fe–N–C SAC) to reduce and even eliminate the stability issue, as MnOx accelerates the degradation of the H2O2 by-product via a disproportionation reaction to weaken the Fenton reaction. As a result, the Mn–Fe–N–C SAC shows an ultralow H2O2 yield and a negligible half-wave potential shift after 10 000 continuous potential cycles, demonstrating excellent ORR stability. Besides, the Mn–Fe–N–C SAC also shows an improved ORR activity compared to the common Fe–N–C SAC. Results show that the MnOx interacts with the Fe–Nx site, possibly forming Fe–Mn or Fe–O–Mn bonds, and enhances the intrinsic activity of single iron sites. This work provides a method to overcome the stability problem of Fe–N–C SACs while still yielding excellent catalytic activity, thus showing great promise for application in PEMFCs.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

