
Tailoring intermolecular interactions in ion gels with rationally designed phosphonic acid polymers†

Abstract
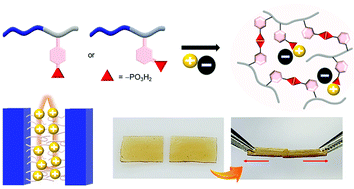
Ideal ion gels exhibit high ionic conductivities and mechanical strengths, but developing such ion gels is challenging, as a high ionic conductivity generally requires a high ionic liquid content, which inevitably reduces the mechanical strength. Herein, we report the controlled syntheses of poly(4-styrenephosphonic acid) (PS4P) and poly(3-styrenephosphonic acid) (PS3P) polymers to achieve challenging goals via the modulation of intermolecular interactions in ion gels containing ionic liquids. Less steric hindrance and increased rotational freedom in PS4P enabled effective swelling and facilitated self-healing of the PS4P-based block copolymer ion gels via robust hydrogen bond formation. Meanwhile, the adjacent chain packing with restricted swelling of the PS3P-based ion gels enabled high toughness and concerted proton hopping. To form dual-crosslinked ion gels, the type of ionic liquid anion was changed to promote ionic interactions, which yielded elastic gels with improved conductivities. Varying the types of ionophobic blocks used in the syntheses of the phosphonated block copolymers allowed the control of the chain stretching in ionic domains. This determined the local ion concentration, thereby modifying the ionic conductivities of the ion gels. Our approach establishes the design of phosphonic acid polymers that exhibit a configurable balance of ionic and hydrogen bonding interactions at the molecular level, which should be a platform for developing advanced ion gels.
- This article is part of the themed collections: Polymer Chemistry Recent HOT Articles and Polymer Networks
 Please wait while we load your content...
Please wait while we load your content...

