
Self-strengthening stimuli-responsive nanocomposite hydrogels†
 ‡b
Jinhye
Bae
*abc
‡b
Jinhye
Bae
*abc
Abstract
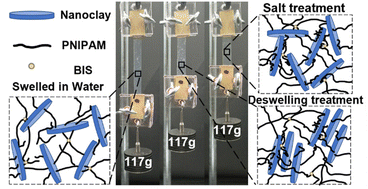
Stimuli-responsive hydrogels with self-strengthening properties are promising for the use of autonomous growth and adaptation systems to the surrounding environments by mimicking biological materials. However, conventional stimuli-responsive hydrogels require structural destruction to initiate mechanochemical reactions to grow new polymeric networks and strengthen themselves. Here we report continuous self-strengthening of a nanocomposite hydrogel composed of poly(N-isopropylacrylamide) (PNIPAM) and nanoclay (NC) by using external stimuli such as heat and ionic strength. The internal structures of the NC-PNIPAM hydrogel are rearranged through the swelling–deswelling cycles or immersing in a salt solution, thus its mechanical properties are significantly improved. The effects of concentration of NC in hydrogels, number of swelling–deswelling cycles, and presence of salt in the surrounding environment on the mechanical properties of hydrogels are characterized by nanoindentation and tensile tests. The self-strengthening mechanical performance of the hydrogels is demonstrated by the loading ability. This work may offer promise for applications such as artificial muscles and soft robotics.
- This article is part of the themed collections: Nanoscale 2023 Emerging Investigators and Celebrating International Women’s Day: Women in Nanoscience
 Please wait while we load your content...
Please wait while we load your content...

