
Biomass derived self-assembled DNA-dot hydrogels for enhanced bacterial annihilation†
 *a
*a
Abstract
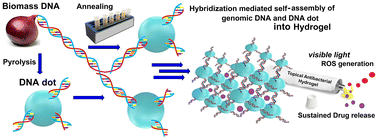
Nanotization of biomass for interesting biomedical applications is still in the nascent stage with no visible market available products. While products derived from biomass DNA and protein have unquestionable biocompatibility, induction of desired properties needs careful manipulation of the biomolecules. Herein, for the first time, we report the transformation of onion derived biomass DNA into DNA-dots through its partial hydrothermal pyrolysis to induce improved mechanical and photophysical properties. The DNA-dots were further used as crosslinkers to create a hydrogel through hybridization-mediated self-assembly with untransformed genomic DNA. The DNA dot–DNA hydrogel sustainably delivers the ciprofloxacin antibiotic as well as produces on-demand reactive oxygen species (ROS) with visible light irradiation. This prompted us to explore the hydrogel as a topical formulation for combination antibiotic Antibacterial-Photodynamic Therapy (APDT) applications. Remarkable annihilation of E. coli and S. aureus, and most importantly two drug-resistant strains of E. coli, shows the success of our sustainable approach.
- This article is part of the themed collection: Emerging concepts in nucleic acids: structures, functions and applications
 Please wait while we load your content...
Please wait while we load your content...

