
Facilitated transport membrane with functionalized ionic liquid carriers for CO2/N2, CO2/O2, and CO2/air separations†
 a
Burcu
Gurkan
*a
a
Burcu
Gurkan
*a
Abstract
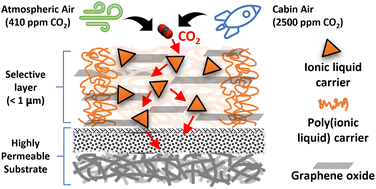
CO2 separations from cabin air and the atmospheric air are challenged by the very low partial pressures of CO2. In this study, a facilitated transport membrane (FTM) is developed to separate CO2 from air using functionalized ionic liquid (IL) and poly(ionic liquid) (PIL) carriers. A highly permeable bicontinuous structured poly(ethersulfone)/poly(ethylene terephthalate) (bPES/PET) substrate is used to support the PIL–IL impregnated graphene oxide thin film. The CO2 separation performance was tested under a mixture feed of CO2/N2/O2/H2O. Under 410 ppm of CO2 at 1 atm feed gas, CO2 permanence of 3923 GPU, and CO2/N2 and CO2/O2 selectivities of 1200 and 300, respectively, are achieved with helium sweeping on the permeate side. For increased transmembrane pressure (>0 atm), a thicker PIL–IL/GO layer was shown to provide mechanical strength and prevent leaching of the mobile carrier. CO2 binding to the carriers, ion diffusivities, and the glass transition temperature of the PIL–IL gels were examined to determine the membrane composition and rationalize the superior separation performance obtained. This report represents the first FTM study with PIL–IL carriers for CO2 separation from air.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

