
Grazing incidence X-Ray diffraction: identifying the dominant facet in copper foams that electrocatalyze the reduction of carbon dioxide to formate†
 bc
and
G. Tayhas R.
Palmore
*ab
bc
and
G. Tayhas R.
Palmore
*ab
Abstract
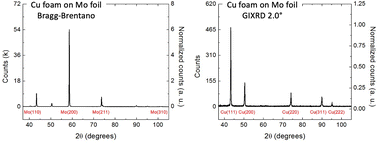
Copper foams have been shown to electrocatalyze the carbon dioxide reduction reaction (CO2RR) to formate (HCOO−) with significant faradaic efficiency (FE) at low overpotentials. Unlike the CO2RR electrocatalyzed at copper foils, the CO2RR electrocatalyzed at porous copper foams selects for HCOO− essentially to the exclusion of hydrocarbon products. Formate is an environmentally friendly organic acid with many applications such as food preservation, textile processing, de-icing, and fuel in fuel cells. Thus, HCOO− is an attractive product from the CO2RR if it is produced at an overpotential lower than that at other electrocatalysts. In this study, grazing incidence X-ray diffraction (GIXRD) was used to identify the dominant surface facet of porous copper foams that accounts for its selectivity for HCOO− during the CO2RR. Included are data from the CO2RR at different temperatures using copper foams as the electrocatalyst. Under optimal reaction conditions at 2 °C, the FE for converting CO2 to HCOO− at Cu foams approaches 50% while the FE for hydrogen gas (H2) falls below 40%, a significant departure from that obtained at polycrystalline Cu foils. Computational studies by others have proposed (200) and (111) facets of Cu foils thermodynamically favour methane and other hydrocarbons, CO, HCOO− from the CO2RR. Results from the GIXRD studies indicate Cu foams are dominated by the (111) facet, which accounts for the selectivity of Cu foams toward HCOO− regardless of temperature used for the CO2RR.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

