
All-carbon microporous graphitic photocatalyst-promoted reduction of CO2 to CO in the absence of metals or dopant elements†

Abstract
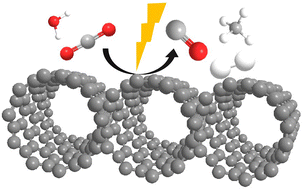
Microporous graphitic carbon (mp-C) derived from the pyrolysis of α-, β-, and γ-cyclodextrins exhibited photocatalytic activity in CO2-saturated acetonitrile–water upon irradiation with UV-Vis light and in the presence of triethanolamine, forming H2 (19 μmol h−1) and CO (23 μmol h−1) accompanied by a lesser proportion of CH4 (4 μmol h−1). The most efficient was the mp-C material derived from α-cyclodextrin (mp-Cα) and having a pore dimension of 0.68 nm. The process also occured, although to a much lesser extent, under simulated sunlight or with UV-Vis irradiation in the absence of a sacrificial agent, with H2O being the electron donor. The origin of the CO was proved by isotopic 13C labelling experiments. Photocurrent measurements proved the occurrence of charge separation and the increase in photocurrent intensity in the presence of CO2. Transient absorption spectroscopy was used to detect the charge separate state decay in the microsecond time scale and proved that a fraction of the photogenerated electrons were able to react with CO2.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

