
Synergistic effect of Cu and Fe small nanoparticles supported on porous N-doped graphitic framework for selective electrochemical CO2 reduction at low overpotential†

Abstract
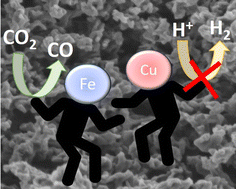
Electrochemical CO2 reduction is an appealing approach to diminish CO2 emissions, while obtaining valuable chemicals and fuels from renewable electricity. However, efficient electrocatalysts exhibiting high selectivity and low operating potentials are still needed. Herein it is reported that Cu and Fe nanoparticles supported on porous N-doped graphitic carbon matrix are efficient and selective electrocatalysts for CO2 reduction to CO at low overpotentials. XRD and Raman spectroscopy confirmed independent Cu and Fe metals as the main phases. HRSEM and HRTEM images show the coral-like morphology of the porous N-doped graphitic carbon matrix supporting Cu and Fe metal nanoparticles (about 10 wt%) homogeneously distributed with an average size of 1.5 nm and narrow size distribution. At the optimum Fe/Cu ratio of 2, this material present high activity for CO2 reduction to CO at −0.3 V vs. RHE with a faradaic efficiency of 96%. Moreover, at −0.5 V vs. RHE this electrocatalyst produces 27.8 mmol of CO gcat−1 h−1, the production rate being stable for 17 h. A synergy between Cu and Fe nanoparticles due to their close proximity in comparison with independent Cu or Fe electrocatalysts was observed.
- This article is part of the themed collection: CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

