
Copper metal organic framework as natural oxidase mimic for effective killing of Gram-negative and Gram-positive bacteria†
 *b
*b
Abstract
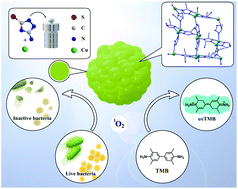
Nanozymes have been widely studied as substitutes for natural enzymes. However, the delicacy of their structures and their unclear catalytic sites make it difficult to maintain their structural robustness and catalytic durability. By mimicking active catalytic sites of natural enzymes and combining them with distinct channels of metal organic frameworks (MOFs), an active copper mimetic oxidase enzyme (Cu-MOF) was designed and synthesized with good structure and clear catalytic sites for improvement in catalytic activity. The Cu-MOFs showed excellent oxidase-like activity with a low Km of 1.09 mM and exogenous ROS generation capacity. The Cu-MOFs exhibited antibacterial efficacy at a low concentration of 12.5 μg mL−1 by an oxidative stress response. These Cu-MOFs with their simple design and effective oxidase mimicking show attractive application prospects in the field of antibacterial and enzyme catalysis.
- This article is part of the themed collection: Celebrating International Women’s day 2024: Women in Nanoscience
 Please wait while we load your content...
Please wait while we load your content...

