
In situ probing the dynamic reconstruction of copper–zinc electrocatalysts for CO2 reduction†
 a
Hao Ming
Chen
*ae
a
Hao Ming
Chen
*ae
Abstract
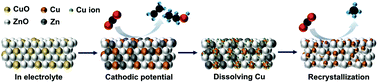
Unravelling the dynamic characterization of electrocatalysts during the electrochemical CO2 reduction reaction (CO2RR) is a critical factor to improve the production efficiency and selectivity, since most pre-electrocatalysts undergo structural reconstruction and surface rearrangement under working conditions. Herein, a series of pre-electrocatalysts including CuO, ZnO and two different ratios of CuO/ZnO were systematically designed by a sputtering process to clarify the correlation of the dynamic characterization of Cu sites in the presence of Zn/ZnO and the product profile. The evidence provided by in situ X-ray absorption spectroscopy (XAS) indicated that appropriate Zn/ZnO levels could induce a variation in the coordination number of Cu sites via reversing Ostwald ripening. Specifically, the recrystallized Cu site with a lower coordination number exhibited a preferential production of methane (CH4). More importantly, our findings provide a promising approach for the efficient production of CH4 by in situ reconstructing Cu-based binary electrocatalysts.
- This article is part of the themed collections: CO2 capture and conversion and 2022 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

