
Boosting photocatalytic CO2 reduction via Schottky junction with ZnCr layered double hydroxide nanoflakes aggregated on 2D Ti3C2Tx cocatalyst†

Abstract
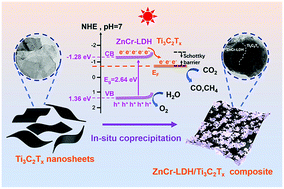
Designing efficient photocatalysts is vital for the photoreduction of CO2 to produce solar fuels, helping to alleviate issues of fossil fuel depletion and global warming. In this work, a novel ZnCr-LDH/Ti3C2Tx Schottky junction is successfully synthesized using an in situ coprecipitation method. ZnCr-LDH nanoflakes collectively grow on the surface of Ti3C2Tx MXene nanosheets. When using Ti3C2Tx MXene as a cocatalyst in the prepared heterojunction, the light absorption intensity, photo-induced electron separation and migration efficiency increase. As a result, the composite ZnCr-LDH/Ti3C2Tx results in significant improvement in the performance of photocatalytic CO2 reduction under simulated solar irradiation. The optimized sample ZCTC25 has the highest photocatalytic CO2 reduction rates of 122.45 μmol g−1 CO and 19.95 μmol g−1 CH4 (after 6 h of irradiation). These values are approximately 2.65 times higher than those of pristine ZnCr-LDH. The product selectivity towards CO is 86%. This work provides a new method for the construction of novel 2D semiconductor photocatalysts and enriches the application of an unusual type of layered double hydroxides in the photoreduction of CO2.
- This article is part of the themed collections: CO2 capture and conversion and 2022 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

