
Tuning the magnetic properties of NiPS3 through organic-ion intercalation†
 a
Yaiza
Asensio,
a
Mihail
Ipatov,
b
Alexander M.
Bittner,
ac
Maider
Ormaza,
*d
Beatriz
Martín-García,
*a
Luis E.
Hueso
ac
and
Marco
Gobbi
*ace
a
Yaiza
Asensio,
a
Mihail
Ipatov,
b
Alexander M.
Bittner,
ac
Maider
Ormaza,
*d
Beatriz
Martín-García,
*a
Luis E.
Hueso
ac
and
Marco
Gobbi
*ace
Abstract
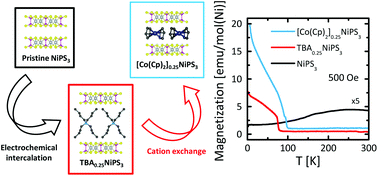
Atomically thin van der Waals magnetic crystals are characterized by tunable magnetic properties related to their low dimensionality. While electrostatic gating has been used to tailor their magnetic response, chemical approaches like intercalation remain largely unexplored. Here, we demonstrate the manipulation of the magnetism in the van der Waals antiferromagnet NiPS3 through the intercalation of different organic cations, inserted using an engineered two-step process. First, the electrochemical intercalation of tetrabutylammonium cations (TBA+) results in a ferrimagnetic hybrid compound displaying a transition temperature of 78 K, and characterized by a hysteretic behavior with finite remanence and coercivity. Then, TBA+ cations are replaced by cobaltocenium via an ion-exchange process, yielding a ferrimagnetic phase with higher transition temperature (98 K) and higher remanent magnetization. Importantly, we demonstrate that the intercalation and cation exchange processes can be carried out in bulk crystals and few-layer flakes, opening the way to the integration of intercalated magnetic materials in devices.
- This article is part of the themed collections: 2025 Journal of Materials Chemistry Lectureship runners-up: Xiaoli Liu & Beatriz Martin Garcia and Nanoscale 2022 Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

