
Computational design of double transition metal MXenes with intrinsic magnetic properties†

Abstract
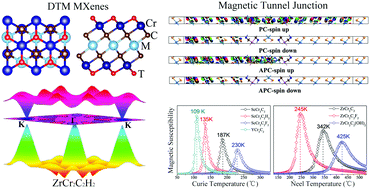
Two-dimensional transition metal carbides (MXenes) have great potential to achieve intrinsic magnetism due to their available chemical and structural diversity. In this work, by spin-polarized density functional theory calculations, we designed and comprehensively investigated 50 double transition metal (DTM) MXenes MCr2CTx (T = H, O, F, OH, or bare) based on the chemical formula of M2C (M = Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Mo, W). We highlight that ferromagnetic half-metallicity, antiferromagnetic semiconduction, as well as antiferromagnetic half-metallicity have been achieved in the DTM MXenes. Herein, ferromagnetic half-metallic ScCr2C2, ScCr2C2H2, ScCr2C2F2, and YCr2C2H2 are characterized with wide band gaps and high Curie temperatures. Very interestingly, the ScCr2C2-based magnetic tunnel junction presents a tunnel magnetoresistance ratio as high as 176 000%. In addition, the antiferromagnetic semiconducting TiCr2C2, ZrCr2C2, and ZrCr2C2(OH)2, possessing moderate band gaps and high Néel temperatures, have been predicted. Especially, the Néel temperature of ZrCr2C2(OH)2 can reach 425 K. Moreover, the Dirac cone-like band structure feature is highlighted in antiferromagnetic half-metallic ZrCr2C2H2. Our study provides a new potential strategy for designing MXenes in spintronics.
- This article is part of the themed collections: Nanoscale Horizons 10th anniversary regional spotlight collection: China, Celebrating International Women’s Day: Women in Nanoscience and Nanoscale Horizons 2023 Lunar New Year Collection
 Please wait while we load your content...
Please wait while we load your content...

