
A self-amplifying nanodrug to manipulate the Janus-faced nature of ferroptosis for tumor therapy†
 *a
*a
Abstract
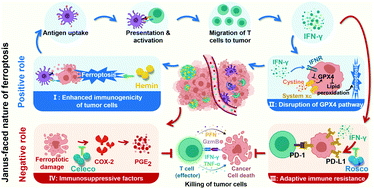
Ferroptosis, an unusual non-apoptotic cell death caused by the iron-dependent accumulation of lipid peroxide, enables the flexible design of an antitumor platform. Specifically, as a positive role, ferroptosis can induce an immune response accompanied with the interferon-γ (IFN-γ)-triggered disruption of the glutathione peroxidase 4 pathway for cascade enhancement of ferroptotic cell death and ferroptosis-induced immunotherapeutic efficacy. However, as a negative role, ferroptosis also triggers inflammation-associated immunosuppression by up-regulation of the cyclooxygenase-2/prostaglandin E2 pathway and IFN-γ-associated adaptive immune resistance by up-regulation of programmed death ligand-1 (PD-L1), impeding the antitumor efficacy of multiple immune cells by immune escape. Negative and positive roles endow ferroptosis with a Janus-faced nature. It is urgent to manipulate the Janus-faced nature of ferroptosis for eliciting the maximized ferroptotic therapeutic efficacy. Herein, a self-amplifying nanodrug (RCH NPs) was designed by co-assembling hemin (ferric porphyrin), celecoxib (anti-inflammatory drug) and roscovitine (cyclin-dependent kinase 5 inhibitor) with the assistance of human serum albumin for reprograming the Janus-faced nature of ferroptosis. During hemin-triggered ferroptosis, celecoxib disrupted the inflammation-related immunosuppression while roscovitine destroyed the IFN-γ-induced up-regulation of PD-L1 via the genetic blockade effect. The RCH NPs thus demonstrated superior therapeutic effects on tumors, thanks to self-amplifying ferroptotic immunotherapy. Our work offers a conceptually innovative strategy for harnessing ferroptosis against tumors.
- This article is part of the themed collections: Nanoscale Horizons 10th anniversary regional spotlight collection: China, Celebrating International Women’s Day: Women in Nanoscience, Nanoscale and Nanoscale Horizons: Nanobiotechnology and Nanomedicine and Nanoscale Horizons Most Popular 2022 Articles
 Please wait while we load your content...
Please wait while we load your content...

