
Environment tolerant, adaptable and stretchable organohydrogels: preparation, optimization, and applications

Abstract
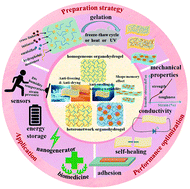
Multiple stretchable materials have been successively developed and applied to wearable devices, soft robotics, and tissue engineering. Organohydrogels are currently being widely studied and formed by dispersing immiscible hydrophilic/hydrophobic polymer networks or only hydrophilic polymer networks in an organic/water solvent system. In particular, they can not only inherit and carry forward the merits of hydrogels, but also have some unique advantageous features, such as anti-freezing and water retention abilities, solvent resistance, adjustable surface wettability, and shape memory effect, which are conducive to the wide environmental adaptability and intelligent applications. This review first summarizes the structure, preparation strategy, and unique advantages of the reported organohydrogels. Furthermore, organohydrogels can be optimized for electro-mechanical properties or endowed with various functionalities by adding or modifying various functional components owing to their modifiability. Correspondingly, different optimization strategies, mechanisms, and advanced developments are described in detail, mainly involving the mechanical properties, conductivity, adhesion, self-healing properties, and antibacterial properties of organohydrogels. Moreover, the applications of organohydrogels in flexible sensors, energy storage devices, nanogenerators, and biomedicine have been summarized, confirming their unlimited potential in future development. Finally, the existing challenges and future prospects of organohydrogels are provided.
- This article is part of the themed collection: 2023 Materials Horizons Lunar New Year collection
 Please wait while we load your content...
Please wait while we load your content...

