
Electromagnetic bioeffects: a multiscale molecular simulation perspective

Abstract
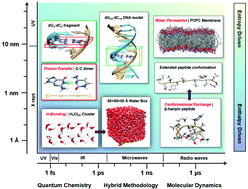
Electromagnetic bioeffects remain an enigma from both the experimental and theoretical perspectives despite the ubiquitous presence of related technologies in contemporary life. Multiscale computational modelling can provide valuable insights into biochemical systems and predict how they will be perturbed by external stimuli. At a microscopic level, it can be used to determine what (sub)molecular scale reactions various stimuli might induce; at a macroscopic level, it can be used to examine how these changes affect dynamic behaviour of essential molecules within the crowded biomolecular milieu in living tissues. In this review, we summarise and evaluate recent computational studies that examined the impact of externally applied electric and electromagnetic fields on biologically relevant molecular systems. First, we briefly outline the various methodological approaches that have been employed to study static and oscillating field effects across different time and length scales. The practical value of such modelling is then illustrated through representative case-studies that showcase the diverse effects of electric and electromagnetic field on the main physiological solvent – water, and the essential biomolecules – DNA, proteins, lipids, as well as some novel biomedically relevant nanomaterials. The implications and relevance of the theoretical multiscale modelling to practical applications in therapeutic medicine are also discussed. Finally, we summarise ongoing challenges and potential opportunities for theoretical modelling to advance the current understanding of electromagnetic bioeffects for their modulation and/or beneficial exploitation in biomedicine and industry.
- This article is part of the themed collections: Showcasing Physical Chemistry research in Australia and New Zealand, 2022 PCCP HOT Articles, New Perspectives on Molecular Simulation of Chemistry and Physics in External Electric Fields and PCCP Perspectives
 Please wait while we load your content...
Please wait while we load your content...

