
mRNA-carrying lipid nanoparticles that induce lysosomal rupture activate NLRP3 inflammasome and reduce mRNA transfection efficiency†
 *abc
*abc
Abstract
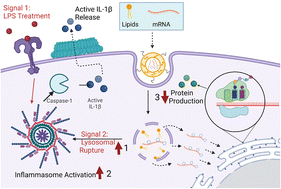
In the last several years, countless developments have been made to engineer more efficient and potent mRNA lipid nanoparticle vaccines, culminating in the rapid development of effective mRNA vaccines against COVID-19. However, despite these advancements and materials approaches, there is still a lack of understanding of the resultant immunogenicity of mRNA lipid nanoparticles. Therefore, a more mechanistic, design-driven approach needs to be taken to determine which biophysical characteristics, especially related to changes in lipid compositions, drive nanoparticle immunogenicity. Here, we synthesized a panel of six mRNA lipid nanoparticle formulations, varying the concentrations of different lipid components and systematically studied their effect on NLRP3 inflammasome activation; a key intracellular protein complex that controls various inflammatory responses. Initial experiments aimed to determine differences in nanoparticle activation of NLRP3 inflammasomes by IL-1β ELISA, which unveiled that nanoparticles with high concentrations of ionizable lipid DLin-MC3-DMA in tandem with high cationic lipid DPTAP and low cholesterol concentration induced the greatest activation of the NLRP3 inflammasome. These results were further corroborated by the measurement of ASC specks indicative of NLRP3 complex assembly, as well as cleaved gasdermin-D and caspase-1 expression indicating complex activation. We also uncovered these activation profiles to be mechanistically correlated primarily with lysosomal rupturing caused by the delayed membrane disruption capabilities of ionizable lipids until the lysosomal stage, as well as by mitochondrial reactive oxygen species (ROS) production and calcium influx for some of the particles. Therefore, we report that the specific, combined effects of each lipid type, most notably ionizable, cationic lipids, and cholesterol, is a crucial mRNA lipid nanoparticle characteristic that varies the endo/lysosomal rupture capabilities of the formulation and activate NLRP3 inflammasomes in a lysosomal rupture dependent manner. These results provide a more concrete understanding of mRNA lipid Nanoparticle-Associated Molecular Patterns for the activation of molecular-level immune responses and provide new lipid composition design considerations for future mRNA-delivery approaches.
- This article is part of the themed collections: Biomaterials Science Emerging Investigator Series, mRNA vaccines against COVID-19: Celebrating the 2023 Nobel Prize in Physiology or Medicine and Biomaterials Science Most Popular 2022
 Please wait while we load your content...
Please wait while we load your content...

