
Short term, low dose alpha-ketoglutarate based polymeric nanoparticles with methotrexate reverse rheumatoid arthritis symptoms in mice and modulate T helper cell responses†

Abstract
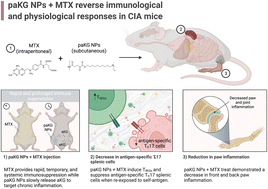
Activated effector T cells induce pro-inflammatory responses in rheumatoid arthritis (RA) which then lead to inflammation of the joints. In this report, we demonstrate that polymeric nanoparticles with alpha keto-glutarate (aKG) in their polymer backbone (termed as paKG NPs) modulate T cell responses in vitro and in vivo. Impressively, a low dose of only three administrations of methotrexate, a clinically and chronically administered drug for RA, in conjunction with two doses of paKG NPs, reversed arthritis symptoms in collagen-induced arthritis (CIA) mice. This was further followed by significant decreases in pro-inflammatory antigen-specific T helper type 17 (TH17) responses and a significant increase in anti-inflammatory regulatory T cell (TREG) responses when CIA treated splenic cells were isolated and re-exposed to the CIA self-antigen. Overall, this study supports the concurrent and short term, low dose of paKG NPs and methotrexate for the reversal of RA symptoms.
- This article is part of the themed collection: Biomaterials Science Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

