
Bile acid linked β-glucan nanoparticles for liver specific oral delivery of biologics
 e
Md
Nurunnabi
*adf
e
Md
Nurunnabi
*adf
Abstract
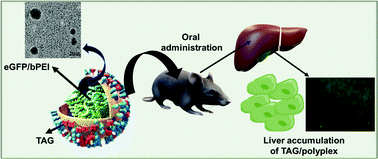
Oral delivery remains one of the most convenient routes for drug administration compared to intravenous, intramuscular, and via suppositories. However, due to the risk of degradation, and proteolysis of molecules in the acidic gastric medium, as well as the difficulty of transporting large molecules through the intestinal membrane, more than half of the therapeutic molecules are prohibited for oral administration. Moreover, most of the large molecules and biological therapeutics are not available in oral dosage form due to their instability in the stomach and inability of intestinal absorption. To achieve expected bioavailability, an orally administered therapeutic molecule must be protected within the stomach, and transportation facilitated via the small intestine. In this project, we have introduced a hybrid carrier, composed of Taurocholic Acid (TA) and β-Glucan (TAG), that is shown to be effective for the simultaneous protection of the biologics in acidic buffer and simulated gastric juice as well as facilitate enhanced absorption and transportation via the small intestine. In this project, we have used an eGFP encoded plasmid as a model biologic to prepare particles mediated with TAG. TAG show the potential of enhancing transfection and expression of eGFP as we have observed two fold higher expression in the cell upon coincubation for 4 h. In vivo studies on orally dosed mice showed that eGFP expression in the liver was significantly higher in TAG containing particles compared to particles without TAG. The findings suggest that the TAG carrier is capable of not only preserving biologics but also transporting them more efficiently to the liver. As a result, this strategy can be employed for a variety of liver-targeted therapeutic delivery to treat a variety of liver diseases.
- This article is part of the themed collection: Biomaterials Science Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

