
Antioxidant Response Activating nanoParticles (ARAPas) localize to atherosclerotic plaque and locally activate the Nrf2 pathway†

Abstract
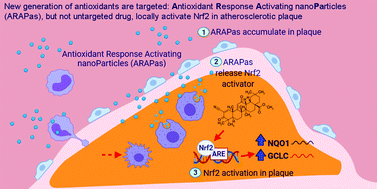
Atherosclerotic disease is the leading cause of death world-wide with few novel therapies available despite the ongoing health burden. Redox dysfunction is a well-established driver of atherosclerotic progression; however, the clinical translation of redox-based therapies is lacking. One of the challenges facing redox-based therapies is their targeted delivery to cellular domains of redox dysregulation. In the current study, we sought to develop Antioxidant Response Activating nanoParticles (ARAPas), encapsulating redox-based interventions, that exploit macrophage biology and the dysfunctional endothelium in order to selectively accumulate in atherosclerotic plaque. We employed flash nanoprecipitation (FNP) to synthesize bio-compatible polymeric nanoparticles encapsulating the hydrophobic Nrf2 activator drug, CDDO-Methyl (CDDOMe-ARAPas). Nuclear factor erythroid 2-related factor 2 (Nrf2)-activators are a promising class of redox-active drug molecules whereby activation of Nrf2 results in the expression of several antioxidant and cyto-protective enzymes that can be athero-protective. In this study, we characterize the physicochemical properties of CDDOMe-ARAPas as well as confirm their in vitro internalization by murine macrophages. Drug release of CDDOMe was determined by Nrf2-driven GFP fluorescence. Moreover, we show that these CDDOMe-ARAPas exert anti-inflammatory effects in classically activated macrophages. Finally, we show that CDDOMe-ARAPas selectively accumulate in atherosclerotic plaque of two widely-used murine models of atherosclerosis: ApoE−/− and LDLr−/− mice, and are capable of increasing gene expression of Nrf2-transcriptional targets in the atherosclerotic aortic arch. Future work will assess the therapeutic efficacy of intra-plaque Nrf2 activation with CDDOMe-ARAPas to inhibit atherosclerotic plaque progression. Overall, our present studies underline that targeting of atherosclerotic plaque is an effective means to enhance delivery of redox-based interventions.
- This article is part of the themed collection: Biomaterials Science Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

