
Designing an improved transition metal phosphide catalyst for hydrogen evolution using experimental and theoretical trends†
Abstract
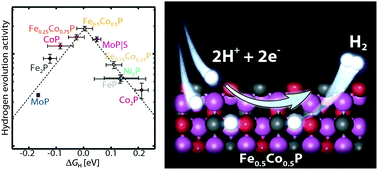
Transition metal phosphides (TMPs) have emerged as highly active catalysts for the hydrogen evolution reaction (HER). However, insights into the trends and limitations in their activity are lacking, and there are presently no guidelines for systematically improving their intrinsic activity. The complexity and variations in their structures further pose challenges in theoretically estimating their activity. Herein, we demonstrate a combined experimental–theoretical approach: by synthesizing different TMPs and comparing experimentally determined HER activities with the hydrogen adsorption free energies, ΔGH, calculated by density functional theory, we determine the level of detail needed in the simulations to bring out useful trends in the experimental data. In particular, we show that the TMPs follow the HER volcano relationship. Using our combined experimental–theoretical model, we predict that the mixed metal TMP, Fe0.5Co0.5P, should have a near-optimal ΔGH. We synthesized several mixtures of Co and Fe phosphides alloys and confirmed that Fe0.5Co0.5P exhibits the highest HER activity of the investigated TMPs. Furthermore, our results suggest that there could be inherent limitations in the intrinsic HER activity of TMPs that prevent them from performing as well as Pt-group metals. Our work demonstrates that it is possible to generate and verify a model of activity trends with predictive capabilities even for new transition metal compounds with varied structures and surface terminations. The identification of an improved mixed metal TMP based on theoretical predictions and subsequent synthesis and testing demonstrates the need for an approach that combines theory and experiment to understand and ultimately design advanced catalysts.
 Please wait while we load your content...
Please wait while we load your content...

