
Single-molecule conductance variations of up to four orders of magnitude via contacting electrodes with different anchoring sites†‡
 a
Wenjing
Hong
*a
a
Wenjing
Hong
*a
Abstract
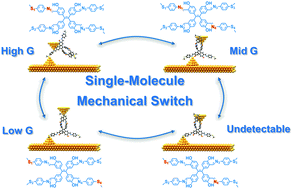
Control of conductance through a single molecule via alternating anchoring points provides a unique perspective to design single-molecule electronic devices. A high conductance difference among different states is essential for a single-molecule electronic device, which is challenging due to restriction of the structural changes in single-molecule junctions. Here, tetraphenylethylene derivatives with multiple anchoring sites are designed and synthesized to investigate single-molecule conductance using the scanning tunneling microscope break junction (STM-BJ) technique. In the break-junction cycle process, connection conformations with different anchor sites have been achieved reversibly to switch single-molecule conductance using the STM-BJ technique. Through mechanical control, these multi-anchored single-molecule junctions can achieve up to three conductance switching cycles, and the variation in conductance reaches up to four orders of magnitude. Theoretical calculations reveal that the conductance change originates from the different connecting sites and the different anchored configurations of single-molecule junctions provide a significantly different transmission. Our findings provide a reliable strategy to manipulate the mechanical switching and shed light on investigating the intrinsic charge transport of multi-anchored molecules.
- This article is part of the themed collections: Bridging excellence —20 years of CCS-RSC collaboration and the Young Chemist Award and Special issue in honour of Kees Hummelen
 Please wait while we load your content...
Please wait while we load your content...

