
Multi length scale porosity as a playground for organic thermoelectric applications
 b
and
Laure
Biniek
*a
b
and
Laure
Biniek
*a
Abstract
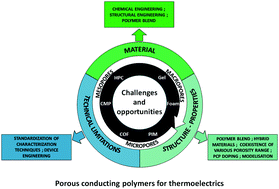
Porous organic materials have interesting materials properties governed not only by their covalent structure but also by their intrinsic porosity which when controlled over multiple length scales gives rise to micro-, meso- and macroporous materials. These materials have been exploited for many years in applications such as gas storage, filtration/separation membranes, or support for catalysts. More recently, porous materials have attracted significant attention as potential harvesters of the abundant waste heat generated in today's society. Taking advantage of the thermoelectric effect, whereupon a temperature gradient is converted to electric voltage, thermoelectric materials and their associated applications are well-suited for this endeavor. Efficient thermoelectric materials must combine a high electrical conductivity and a low thermal conductivity. Since in porous materials, these properties can potentially be optimized independently, they are intriguing candidates for further exploration. Here, we give an overview of the different classes of porous conducting polymers (PCPs) and provide a thorough survey of their recent use in the broader context of thermoelectrics. We also aim to identify the major challenges and future perspectives for porous organic thermoelectric materials.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry Lectureship runners-up: Kwabena Bediako and Laure Biniek and 2022 Journal of Materials Chemistry Lectureship shortlisted candidates
 Please wait while we load your content...
Please wait while we load your content...

