
Dysprosium–dianthracene framework showing thermo-responsive magnetic and luminescence properties†
 *a
Li-Min
Zheng
*a
*a
Li-Min
Zheng
*a
Abstract
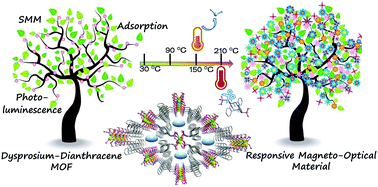
Metal–organic frameworks showing stimuli-responsive magneto-optical properties are of intensive current interest for potential applications in spintronics and molecular devices. By incorporating a thermo-responsive dianthracene component into a lanthanide phosphonate framework, we successfully obtained compounds Ln2(amp2H2)2(C2O4)(H2O)2·xH2O (MDAF-4Ln, Ln = Dy, x = 10; Gd, x = 9; Er, x = 7; and amp2H4 = pre-photodimerized 9-anthrylmethylphosphonic acid). These compounds are isostructural showing three-dimensional open framework structures composed of Ln-chains and dianthracence linkages. Within the chain, the dimers of {Ln2O2(OPO)2} are connected by oxalate anions. The MOFs generate channels along the a-axis which are hydrophilic lined with protonated phosphonate moieties, and hence exhibit high affinity towards adsorption of water and ammonia. Thermogravimetric analysis and differential scanning calorimetry (DSC) measurements revealed that MDAF-4Ln experienced two consecutive phase transitions: first, the loss of all lattice and coordinated water molecules and then, the dissociation of amp2H22−. Detailed studies on MDAF-4Dy demonstrated that the thermo-induced structural transformations were accompanied by significant changes in its single-molecule magnetic behavior and luminescence properties.
- This article is part of the themed collection: Materials for molecular electronics and magnetism
 Please wait while we load your content...
Please wait while we load your content...

