
Highly sensitive non-enzymatic electrochemical glucose sensor surpassing water oxidation interference†
 *a
and
Tharamani C.
Nagaiah
*a
*a
and
Tharamani C.
Nagaiah
*a
Abstract
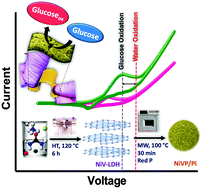
An electrochemical non-enzymatic sensor based on a NiVP/Pi material was developed for the selective and sensitive determination of glucose. The novel sensor showed a high sensitivity of 6.04 mA μM−1 cm−2 with a lowest detection limit of 3.7 nM in a wide detection range of 100 nM–10 mM. The proposed sensor exhibited a superior selectivity without any interference from the oxygen evolution reaction during glucose sensing. We also found that this glucose sensor showed negligible interference from various interferents, such as ascorbic acid, uric acid, dopamine and sodium chloride. Additionally, a novel flexible sensor was developed by coating the NiVP/Pi over Whatman filter paper, which exhibited two linear ranges of 100 nM to 1 μM and 100 μM to 10 mM with an ultra-sensitivity of 1.130 mA μM−1 cm−2 and 0.746 mA μM−1 cm−2, respectively, in 0.1 M NaOH. The proposed sensor was tested with human blood serum samples demonstrating its practical application. Our findings provide a new route by fine tuning the composition of nickel and vanadium that sheds new light on better understanding the processes. This NiVP/Pi-based sensor offers a new approach towards the electrochemical detection of glucose, enabling glucose monitoring in a convenient way.
- This article is part of the themed collections: Celebrating International Women’s Day: Women in Materials Science and 2021 Journal of Materials Chemistry B most popular articles
 Please wait while we load your content...
Please wait while we load your content...

