
Delivery of a system xc− inhibitor by a redox-responsive levodopa prodrug nanoassembly for combination ferrotherapy†

Abstract
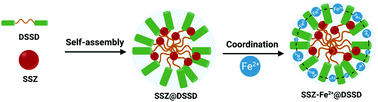
A comprehensive understanding of ferroptosis signaling pathways significantly contributes to the advances in cancer ferrotherapy. Herein, we constructed a self-assembled prodrug nanosystem targeting system xc−, a key regulator for ferroptosis, to amplify the therapeutic efficacy of cancer ferrotherapy. The prodrug nanosystem is assembled between sulfasalazine (SSZ, a ferroptosis resistance inhibitor) and disulfide-bridged levodopa (DSSD) that can chelate Fe2+ ions to form SSZ–Fe2+@DSSD, and the resulting nanoassembly can not only inhibit ferroptosis resistance, but also generate ROS in the tumor microenvironment. Whereas the prodrug nanosystem is stable in the physiological environment, it becomes unstable in the tumoral and intracellular reductive microenvironment, where the disulfide linkers are disrupted by high levels of glutathione (GSH), triggering the release of active Fe2+ and SSZ. Under the Fenton reaction, the released Fe2+ thus can induce ferroptosis, which is amplified by SSZ-mediated inhibition of ferroptosis resistance to synergistically improve the therapeutic efficacy of ferroptosis. Our study thus provides an innovative prodrug strategy to advance anticancer ferroptosis.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

