
The crucial roles of the configurations and electronic properties of organic hole-transporting molecules to the photovoltaic performance of perovskite solar cells
 *a
and
Zhen
Li
*ab
*a
and
Zhen
Li
*ab
Abstract
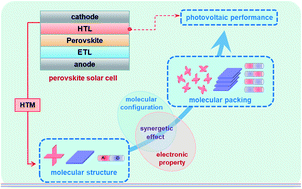
Perovskite solar cells have become one of the most promising technologies to make use of solar energy; to date, the power conversion efficiencies (PCEs) have been improved from 3.8% to 25.6%. Hole-transporting materials (HTMs) play an important role in the photovoltaic conversion process by extracting photogenerated holes and transporting charges. However, the relationship among the molecular structure of HTMs, molecular packing in hole-transporting layers (HTLs) and the device performance of perovskite solar cells (PSCs) has not been explained systematically. In this review, the structure–property relationship of HTMs is discussed from the aspects of molecular configuration, electron properties, and their synergetic effects, to provide useful guidance for the HTM design and PSC development.
- This article is part of the themed collection: Editor’s Choice: Perovskite-based solar cells
 Please wait while we load your content...
Please wait while we load your content...

