
Three-in-one: achieving a robust and effective hydrogen-evolving hybrid material by integrating polyoxometalate, a photo-responsive metal–organic framework, and in situ generated Pt nanoparticles†
 *a
*a
Abstract
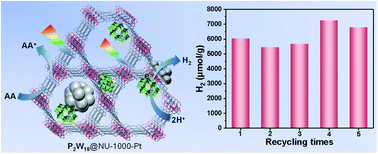
A Wells–Dawson-type polyoxometalate ([P2W18O62]6−, denoted as P2W18), a photo-responsive Zr-based metal–organic framework (MOF, NU-1000), and in situ generated Pt nanoparticles were successfully integrated into a three-in-one hybrid material (P2W18@NU-1000-Pt) using a facile impregnation and subsequent photoreduction method. The resulting three-in-one hybrid material exhibits effective and robust activity towards photocatalytic hydrogen production in a water-compatible system under Xe-lamp irradiation, achieving a hydrogen production of 35 100 μmol g−1 and a turnover number (TON) of 5484 versus moles of Pt after 5 days of reaction. The post-catalysis three-in-one hybrid composite could be easily recycled at least 5 times without declining catalytic activity. Moreover, such a water-compatible photocatalytic system also reveals potential practical applications for catalyzing hydrogen production under natural sunlight irradiation. A possible photocatalytic mechanism was elucidated based on various photophysical and spectroscopic analyses, proving the importance of synergistic cooperation of the NU-1000 MOF, the P2W18 cluster, and in situ generated Pt NPs in such a three-in-one P2W18@NU-1000-Pt hybrid photocatalyst.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

