
Modulating the electronic structure of nanomaterials to enhance polysulfides confinement for advanced lithium–sulfur batteries

Abstract
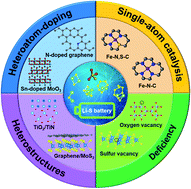
Lithium–sulfur (Li–S) battery is one of the most promising next-generation energy storage systems. Nevertheless, owing to the high solubility of lithium polysulfides (LiPSs) in the ether-based electrolyte, plenty of LiPSs shuttle between cathode and anode under an electric field. This shuttle effect corrodes the lithium metal, and thus causes serious capacity fading, which is regarded as a major barrier to the commercial application of Li–S batteries. Engineering the surficial structure of nanomaterials can promote affinity between cathode and LiPSs, while simultaneously facilitating redox kinetics of LiPSs, leading to a low concentration of LiPSs in electrolyte for restraining the shuttle effect. Herein, we review recent progress in manipulating the electronic structure of nanomaterials featuring high conductivity, strong absorption and catalytic properties for Li–S batteries. We first discuss the mechanism of the conversion of LiPSs with different pathways. Moreover, we showcase the design strategies of nanomaterials with the modulated surface, including heterostructures, deficiency strategy, heteroatom-doping strategy and metal-single-atom catalyst. Future perspectives and challenges are also proposed for constructing stable Li–S batteries with high energy density.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

