
Coacervation-driven instant paintable underwater adhesives with tunable optical and electrochromic properties†

Abstract
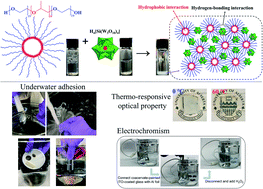
Coacervation generally refers to liquid–liquid phase separation when mixing oppositely charged polyelectrolytes in an aqueous solution, which produces a polyelectrolyte-dense coacervate phase in a dilute solution phase. Coacervation plays a crucial role in many biological processes such as the versatile underwater adhesion of sessile organisms, cellular compartmentalization, and cell replication, which inspires a surge of biomimetic designs of underwater adhesives. However, conventional coacervate-based underwater adhesives are usually weak and lack functionalities, limiting their practical applications in biomedical and engineering fields. Herein, a novel instant underwater adhesive is presented based on coacervation via one-step mixing of aqueous solutions of silicotungstic acid (SiW) and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123) micelles, driven by hydrogen-bonding and hydrophobic interactions. The as-prepared adhesive possesses instant and excellent underwater adhesion (up to 479.6 kPa on poly(methyl methacrylate)) and can be facilely painted underwater on diverse substrates, showing resistance to water flush as well as repeatable stretching and bending of the substrates, superior to many previously reported coacervates. The adhesive also exhibits outstanding stability in high-salt aqueous solutions up to 3 M for at least 1200 h. The introduction of P123 endows the adhesives with thermo-responsive optical properties, while the innate reduction-related color switch of SiW offers electrochromic properties. The functional adhesives hold great promise in bioengineering applications and for the fabrication of novel flexible electronics such as smart aqueous batteries and low-power electrochromic windows.
- This article is part of the themed collection: Polymers in liquid formulations
 Please wait while we load your content...
Please wait while we load your content...

