
Organic electrode materials for non-aqueous, aqueous, and all-solid-state Na-ion batteries

Abstract
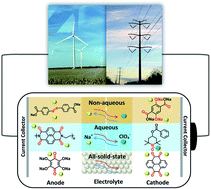
Na-ion batteries (NIBs) are promising alternatives to Li-ion batteries (LIBs) due to the low cost, abundance, and high sustainability of sodium resources. However, the high performance of inorganic electrode materials in LIBs does not extend to NIBs because of the larger ion size of Na+ than Li+ and more complicated electrochemistry. Therefore, it is vital to search for high-performance electrode materials for NIBs. Organic electrode materials (OEMs) with the advantages of high structural tunability and abundant structural diversity show great promise in developing high-performance NIBs. To achieve advanced OEMs for NIBs, a fundamental understanding of the structure–performance correlation is desired for rational structure design and performance optimization. In this review, recent advances in developing OEMs for non-aqueous, aqueous, and all-solid-state NIBs are presented. The challenges, advantages, mechanisms, development, and applications of advanced OEMs in NIBs are also discussed. Perspectives for the innovation of structure design principle and future research direction of OEMs in non-aqueous, aqueous, and all-solid-state NIBs are provided.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

