
Defect formation and amorphization of Zn-MOF-74 crystals by post-synthetic interactions with bidentate adsorbates†

Abstract
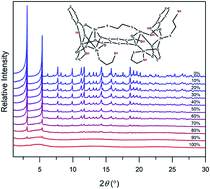
The controlled introduction of defects into MOFs is a powerful strategy to induce new physiochemical properties and improve their performance for target applications. Herein, we present a new strategy for defect formation and amorphization of canonical MOF-74 frameworks based on fine-tuning of adsorbate–framework interactions in the metal congener, hence introducing structural defects. Specifically, we demonstrate that controlled interactions between the MOF and bidentate ligands adsorbed in the pores initiate defect formation and eventual amorphization of the crystal. These structural features unlock properties that are otherwise absent in the ordered framework, such as broad-band fluorescence. The ability to introduce defects by adsorbate–framework interactions, coupled with the inherent tunability and modularity of these structures, provides a new route for the synthesis of diverse heterogeneous and hybrid materials.
- This article is part of the themed collections: 2024 Journal of Materials Chemistry Lectureship runners-up: Maxx Arguilla and Phillip Milner and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

