

Abstract
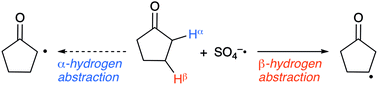
Many hydrogen abstraction reactions on sp3 carbons with oxyradicals take place site-selectively (regioselectively). To investigate this selectivity, ab initio and density functional theory (DFT) calculations were carried out using cyclopentanone and SO4−˙ as the substrate and oxyradical, respectively. At the ωB97XD/6-311+G(d,p) level, the energy barriers for the forward process (ΔE1‡) of both α- and β-hydrogen abstraction were predicted to be 54.6 and 50.9 kJ mol−1, respectively. Consideration of solvent effects (acetonitrile) decreased these energy barriers to 33.2 and 26.1 kJ mol−1, respectively. These calculation outcomes suggested that β-hydrogen abstraction would be favourable, which supports experimental findings (i.e. β-selective abstraction). At the ωB97XD level, investigations into hydrogen abstraction from cyclohexanone with SO4−˙ confirmed the regioselectivity observed experimentally. Hydrogen abstractions from 2-propylpyridine and 3-methyl-1-butanol using SO4−˙, which are unknown reactions, were also calculated using the DFT method, and the predicted regioselectivity was consistent with that in the known reactions using tetrabutylammonium decatungstate (TBADT). In addition, regioselectivities in unexplored hydrogen abstractions of cyclopentanone by several oxyradicals were predicted. Natural bond orbital (NBO) analysis carried out at the ωB97XD level indicated that the transferred hydrogen atom is partially positively charged when abstracted by an oxyradical. Interestingly, hydrogens bonded to the most positively charged carbon in the substrate were predominantly abstracted by oxyradicals in practice, which should be a simple compass for predicting regioselectivity in the functionalisation of C(sp3)–H bonds with oxyradicals.
- This article is part of the themed collection: Mechanistic, computational & physical organic chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

