
Size-controlled nanocrystals reveal spatial dependence and severity of nanoparticle coalescence and Ostwald ripening in sintering phenomena†
 b
and
Matteo
Cargnello
*a
b
and
Matteo
Cargnello
*a
Abstract
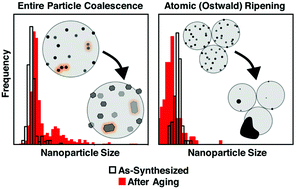
A major aim in the synthesis of nanomaterials is the development of stable materials for high-temperature applications. Although the thermal coarsening of small and active nanocrystals into less active aggregates is universal in material deactivation, the atomic mechanisms governing nanocrystal growth remain elusive. By utilizing colloidally synthesized Pd/SiO2 powder nanocomposites with controlled nanocrystal sizes and spatial arrangements, we unravel the competing contributions of particle coalescence and atomic ripening processes in nanocrystal growth. Through the study of size-controlled nanocrystals, we can uniquely identify the presence of either nanocrystal dimers or smaller nanoclusters, which indicate the relative contributions of these two processes. By controlling and tracking the nanocrystal density, we demonstrate the spatial dependence of nanocrystal coalescence and the spatial independence of Ostwald (atomic) ripening. Overall, we prove that the most significant loss of the nanocrystal surface area is due to high-temperature atomic ripening. This observation is in quantitative agreement with changes in the nanocrystal density produced by simulations of atomic exchange. Using well-defined colloidal materials, we extend our analysis to explain the unusual high-temperature stability of Au/SiO2 materials up to 800 °C.
- This article is part of the themed collections: Nanoscale and Nanoscale Horizons: Nanoparticle Synthesis, Nanoscale 2021 Emerging Investigators and Editor’s Choice: Single-atom and nanocluster catalysis
 Please wait while we load your content...
Please wait while we load your content...

