
Selective visible-light driven highly efficient photocatalytic reduction of CO2 to C2H5OH by two-dimensional Cu2S monolayers†

Abstract
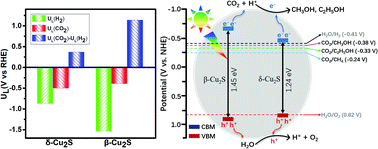
Solar-driven highly-efficient photocatalytic reduction of CO2 into value-added fuels has been regarded as a promising strategy to assuage the current global warming and energy crisis, but developing highly product-selective, long-term stable and low-cost photocatalysts for C2 production remains a grand challenge. Herein, we demonstrate that two-dimensional β- and δ-phase Cu2S monolayers are promising photocatalysts for the reduction of CO2 into C2H5OH. The calculated potential-limiting steps for the CO2 reduction reaction (CO2RR) are less than 0.50 eV, while those for the hydrogen evolution reaction are as high as 1.53 and 0.87 eV. Most strikingly, the C–C coupling only needs to overcome an ultra-low kinetic barrier of ∼0.30 eV, half of that on the Cu surface, indicating that they can boost the C2H5OH conversion efficiency greatly. Besides, these catalysts also exhibit satisfactory band edge positions and suitable visible light absorption, rendering them ideal for the visible light driven CO2RR. Our work not only provides a promising photocatalyst for achieving the efficient and selective CO2RR, but also brings new opportunities for advanced sustainable C2H5OH product.
- This article is part of the themed collections: Nanoscale Horizons 10th anniversary regional spotlight collection: China, Celebrating International Women’s Day: Women in Nanoscience, Nanoscale Horizons 2022 Lunar New Year Collection, Horizons Community Board Collection: Solar Energy Conversion and Nanoscale Horizons Most Popular 2021 Articles
 Please wait while we load your content...
Please wait while we load your content...

