
Toward printable solar cells based on PbX colloidal quantum dot inks

Abstract
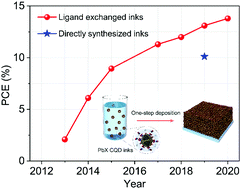
Lead chalcogenide (PbX, X = S, Se) colloidal quantum dots (CQDs) are promising solution-processed semiconductor materials for the construction of low-cost, large-area, and flexible solar cells. The properties of CQDs endow them with advantages in semi-conducting film deposition compared to other solution-processed photovoltaic materials, which is critical for the fabrication of efficient large-area solar cells towards industrialization. However, the development of large-area CQD solar cells is impeded by the conventional solid-state ligand exchange process, where the tedious processing with high expense is indispensable to facilitate charge transport of CQD films for photovoltaic applications. In the past several years, the rapid development of CQD inks has boosted the device performance and dramatically simplified the fabrication process. The CQD inks are compatible with most of the industrialized printing techniques, demonstrating potential in fabricating solar modules for commercialization. This article aims to review the recent advances in solar cells based on PbX CQD inks, including both lab-scale and large-area photovoltaic devices prepared from solution-phase ligand exchange (SPLE) as well as the recently invented “one-step” synthesis. We expect to draw attention to the enormous potential of CQD inks for developing high-efficiency and low-cost large-area photovoltaics.
- This article is part of the themed collection: Nanoscale Horizons 2022 Lunar New Year Collection
 Please wait while we load your content...
Please wait while we load your content...

